
Синдром Вільямса
(Williams Syndrome)
Наталія Григорівна Горовенко
Д-р мед. наук, професор, чл.-кор. АМН України,
Київська медична академія після дипломної освіти ім. П.Л. Шупика
Тетяна Едуардівна Зерова-Любимова
Канд. біолог. наук,
Київська медична академія після дипломної освіти ім. П.Л. Шупика
Наталія Олексіївна Тищенко
Київська медична академія після дипломної освіти ім. П.Л. Шупика
Олена Генадіївна Євсеєнкова
Київська медична академія після дипломної освіти ім. П.Л. Шупика
Рецензент:
Ігор Романович Бариляк
Д-р мед. наук, професор,
Голова проблемної комісії “Медична генетика” МОЗ і АМН України
Перелік умовних скорочень:
FISH – флуоресцентна іn situ гібридизація
МРТ – магнітно-резонансна томографія
Ехо-КГ – ехо-кардіографія
УЗД – ультразвукове дослідження
УФ випромінювання – ультрафіолетове випромінювання
Вступ:
Синдром Вільямса – мультисистемне захворювання, причиною якого є мікроделеція сегменту 7q11.23. Частота синдрому складає 1:20000 новонароджених. Основними клінічними ознаками є дизморфічні риси обличчя, затримка розумового розвитку та труднощі навчання, унікальні психологічні особливості, гіперкальціємія, сполучнотканинні аномалії, що обумовлені дефектом гену еластину ELN. За допомогою флуоресцентної іn situ гібридизації (FISH) ідентифікувати мікрохромосомну аномалію (мікроделецію 7q11.23) можна у 99% пацієнтів з клінічними ознаками синдрому Вільямса. Лабораторне підтвердження діагнозу дає можливість правильно визначити прогноз і тактику ведення хворого, провести медико-генетичне консультування в родині.
В Україні тільки нещодавно з’явилася можливість проведення підтверджуючої лабораторної діагностики синдромів з мікроабераціями хромосом, і зокрема синдрому Вільямса за допомогою флуоресцентної гібридизації in situ (FІSH). Незважаючи на те, що синдром Вільямса має дуже специфічну клінічну картину, знайомі з ним практично лише фахівці з вузькою спеціалізацією – генетики. Трапляються випадки пізньої діагностики синдрому в дорослому віці, коли проведення реабілітаційних медичних та педагогічних заходів вже не дають ефекту. Своєчасна діагностика синдрому та дотримання рекомендованої тактики ведення хворих з синдромом Вільямса дозволяє в багатьох випадках значно поліпшити медичний та, особливо, соціальний прогноз для пацієнтів. Крім того, застосування молекулярно-цитогенетичного тесту дає можливість провести високодостовірну пренатальну діагностику та медико-генетичне консультування в родині.
Методичні рекомендації по клінічній та лабораторній діагностиці синдрому Вільямса, особливостям реабілітації хворих та медико-генетичному консультуванню підготовлені вперше в Україні і базуються на великому практичному досвіді авторів та рекомендаціях провідних генетичних та педіатричних установ світу.
Матеріал, викладений в методичних рекомендаціях, призначений для лікарів-генетиків, неонатологів, педіатрів, кардіологів, кардіохірургів, дитячих неврологів та психіатрів.
Основна частина:
Перший опис синдрому було зроблено Williams JC із співавторами в 1961 році. Роком пізніше, у 1962 році, Beuren AJ із співавторами доповнили спектр фенотипових ознак синдрому, який включав надклапанний стеноз аорти, специфічні риси обличчя та розумову відсталість. Пізніше було виявлено, що більшість пацієнтів мають також ідіопатичну гіперкальціємію. Слід зазначити, що випадки ідіопатичної гіперкальціємії були описані і раніше в Англії після другої світової війни під час епідемії гіперкальціємії новонароджених, яка була наслідком перенасичення дитячого харчування вітаміном Д. Після зниження вмісту вітаміну Д рівень кальцію у більшості дітей нормалізувався, але у деяких пацієнтів гіперкальціємія залишилась, крім цього у них спостерігались незвичайні риси обличчя та затримка розвитку. Цей особливий стан отримав назву синдрома Вільямса – Бойрена, або „обличчя ельфа” (враховуючи специфічність рис обличчя пацієнтів в дитинстві). З введенням в практику флуоресцентної in situ гібридізації було виявлено, що 99% хворих мають мікроделецію сегменту q11.2 хромосоми 7.
Етіологія:
Причиною розвитку синдрому Вільямса є мікроделеція сегменту q11.2 хромосоми 7 розміром ~1,5 Mb, в якому картовано 16 генів, включаючи ген еластину ELN. Саме втрата гену еластину і обумовлює розвиток всієї сукупності сполучнотканинних аномалій, які спостерігаються при цьому синдромі. З’ясовано, що делеція з однаковою частотою має як материнське, так і батьківське походження. Мейотичні помилки саме в цьому сегменті, в так званій гарячій точці, обумовлені наявністю в цій ділянці хромосоми 7 великої кількості повторів тринуклеотидних послідовностей. Чіткого взаємозв’язку між більшістю картованих в критичній ділянці генів та фенотиповими проявами синдрому Вільямса не виявлено, хоча відомо, що гени FZD3, BCL7B, STX1A, LIMK1, та CYLN2 експресуються в нервовій тканині, і тому можуть мати вплив на особливості нервово-психічного розвитку при синдромі Вільямса.
Таблиця 1. Гени, які картовано в ділянці, що делетована при синдромі Вільямса
(Annu. Rev. Genomics Hum. Genet. 2000. 1:461–484)
Клініка:
До головних ознак синдрому Вільямса відносяться: специфічні риси обличчя, сукупність яких дає враження „обличчя ельфа”, вроджені вади серця (найбільш специфічною є надклапанний стеноз аорти), як один з проявів аномалій сполучної тканини, ідіопатична гіперкальціємія та гіперкальціурія на першому році життя, затримка розумового розвитку з формуванням особливого психо-когнітивного профілю. Крім основних симптомів, хворі мають затримку фізичного розвитку та порушення інших органів та систем.
Лицьовий дизморфізм в періоді новонародженості та ранньому дитинстві включає: широкий лоб, бітемпоральне звуження, низьке перенісся, періорбітальна припухлість, зірчаста райдужка, косоокість, короткий ніс або відкриті наперед ніздрі, довгий фільтр, повні губи, широкий рот, повні щоки, порушення прикусу та дрібні зуби, мікрогнатія, виступаюча часточка вушної мушлі. Слід зазначити, що повний спектр перелічених малих аномалій обличчя, який дає враження “обличчя ельфа” та дозволяє поставити клінічний діагноз, формується на 3-4-му році життя. Проте вже після 10-15-річного віку для пацієнтів з синдромом Вільямса характерним є довге обличчя, довга шия, виступаючі надбрівні дуги, пласка середня третина обличчя, широкий рот та повні губи, вузьке перенісся, широкий кінчик носа.
До спектру сполучнотканинних аномалій входять грубий голос, за рахунок змін голосових зв’язок, пахові та пупкові кили, дивертикули кишок або сечового міхура, гіпереластичність шкіри, гіпермобільність суглобів, а також серцево-судинні порушення.
Аномалії серцево-судинної системи присутні більш ніж у 80% хворих. Найбільш частою маніфестацією еластинової артеріопатії є надклапанний стеноз аорти. Також виявляють периферичний стеноз легеневої артерії, пролапс мітрального клапану, артеріальну гіпертензію. З віком ступінь артеріального стенозу збільшується. Розвиток обструкції можливий в будь-якій ділянці артеріального русла. Стенози коронарних судин бувають частою причиною раптової смерті у деяких хворих з синдромом Вільямса. Звуження судин головного мозку можуть спричинити розвиток інсульту. Стенози ниркових артерій – це найбільш часта причина артеріальної гіпертензії, вона присутня у близько 50% пацієнтів. Взагалі, саме серцево-судинні аномалії є основною причиною смерті при синдромі Вільямса.
Ідіопатична гіперкальціємія спостерігається зазвичай протягом перших 18 місяців життя. Високий рівень кальцію крові мають до 15% дітей, високий рівень екскреції кальцію з сечею (гіперкальціурію) мають до 30% дітей на першому році життя. Гіперкальціємія може спричиняти розвиток дратівливості, блювання, закрепу та крампі (болючих посмикувань м’язів). Гіперкальціурія може бути причиною розвитку нефрокальцинозу в дорослому віці. Етіологія порушення метаболізму кальцію при синдромі Вільямса ще не з’ясована.
Особливості психомоторного розвитку. Однією з найчастіших фенотипових ознак синдрому Вільямса є особливості психомоторного розвитку та деякі риси особистості пацієнтів, а саме, надзвичайна дружелюбність пацієнтів при наявності у них середньої та легкої ступені розумової відсталості, яка діагностується у 75% дітей.
Вже з перших років життя відмічається нерівномірна структура порушеного психомоторного розвитку.
- Перше півріччя – спостерігається затримка розвитку інтегративних функцій, які безпосередньо пов’язані з руховим аналізатором, та насамперед зорово-моторною координацією. В подальшому все більше проявляється порушення моторних функцій та координації рухів. Це призводить до специфічних труднощів в оволодінні навичками самообслуговування. Наявність моторної недостатності та відставання в строках розвитку статичних та локомоторних функцій інколи призводить до помилкового встановлення діагнозу дитячого церебрального паралічу.
- Ранній дитячий вік – відзначається значне відставання в темпах мовного розвитку. Згідно деяких спостережень перші слова діти з синдромом Вільямса починають говорити не раніше 2,5-3 років. З віком затримка мовного розвитку змінюється високою мовною активністю, надмірною балакучістю. Діти легко повторюють мову дорослих, багато говорять, але часто недоречно. Спостерігаються порушення вимовляння звуків, причиною є дефекти зубо-щелепної системи та дизартрія. В деяких випадках висока мовна активність, легкість повторювання окремих слів та фраз може вдало маскувати інтелектуальну недостатність.
- Шкільний вік – нерівномірність психічного розвитку проявляє себе у вигляді різної здібності до засвоєння математики та читання. Зазвичай відмічаються труднощі в оволодінні рахуванням, навіть виконання простих математичних дій на конкретних прикладах. Читання дається відносно легко. Порушення у сфері розвитку тонкої моторики обумовлює труднощі в оволодінні письмом. До психологічних проблем хворих з синдромом Вільямса також відносяться порушення уваги з гіперактивністю та тривожність. Розумова відсталість може сполучатися з незрілістю емоційно-вольової сфери. Частина хворих має ознаки лобної недостатності: діти не враховують поточної ситуації, у них відсутня самокритичність, часто спостерігається ейфорія з порушеннями регуляторної діяльності. Діти з синдромом Вільямса, як часто відзначають батьки, мають хороший музичний слух, відчуття ритму і схильні до занять музикою та співом.
- Поведінка дітей з синдромом Вільямса в цілому характеризується надзвичайною дружелюбністю, високою соціальною активністю, чутливістю, легкістю в зав’язуванні нових контактів, лагідністю. Хворі не за віком наївні та дуже безпосередні. Діти легко виконують вказівки дорослих, але їхня особиста діяльність часто неорганізована. Емоційні особливості дітей з синдромом Вільямса обумовлюють прихильність до них оточуючих, особливо дорослих.
Фізичний розвиток при синдромі Вільямса характеризується низьким зростом та малою вагою тіла. Пренатальна затримка росту спостерігається у 50-70% випадків. Причинами затримки росту та повільної прибавки ваги у новонароджених часто є порушення годування, гастроезофагальний рефлюкс, хронічний закреп. До 70% дітей мають зріст нижче середнього показника зросту їхніх батьків. Пубертат настає зазвичай раніше ніж у однолітків, але передпубертатний ростовий стрибок дуже короткий, тому зріст більшості дорослих пацієнтів з синдромом Вільямса нижче 3 центиля.
Неврологічні порушення у дітей з синдромом Вільямса включають гіпотонію, гіперрефлексію, ознаки порушення функції мозочка. В деяких випадках, при проведенні МРТ головного мозку, виявлялось зменшення об’єму мозку зі збереженням нормальних розмірів мозочка, також описані поодинокі випадки наявності мальформації Кіарі І (зміщення мозочка у каудальному напрямку).
Офтальмологічні аномалії включають конвергентну косоокість, гіперметропію, гіпоплазію строми радуйжки, зниження гостроти зору.
Орган слуху. Ще однією особливістю при синдромі Вільямса є гіперчутливість до гучних звуків, яка має місце у пацієнтів будь-якого віку. У дітей молодшого віку часто розвивається хронічний отит.
Порушення функції травної системи включають порушення годування у вигляді зригувань, рефлюксну хворобу, закріп (до 40%) та дивертикульоз кишок. Причинами хронічного абдомінального болю у дорослих із синдромом Вільямса можуть бути виразкова хвороба, жовчокам’яна хвороба, дивертикульоз, хронічний закреп або кила стравохідного отвору діафрагми.
Аномалії сечовивідної системи при синдромі Вільямса включають вади розвитку нирок, дивертикули сечового міхура, нефрокальциноз. До 50% дітей з синдромом Вільямса страждають на енурез, до 30% дорослих хворих мають хронічні інфекційні захворювання сечових шляхів.
До вад розвитку м’язової-скелетної системи, що описані у хворих з синдромом Вільямса, відносять радіоульнарний синостоз, кіфоз, лордоз та сколіоз. У дітей часто спостерігається гіпермобільність суглобів, але в більш дорослому віці формуються контрактури. Аномалії суглобів в комбінації з постуральними порушенням призводять до формування порушень ходи.
Діагностика синдрому Вільямса:
Діагноз синдрому Вільямса встановлюється при наявності достатньої кількості характерних клінічних ознак (табл. 3) (клінічний діагноз) та підтверджується молекулярно-цитогенетичним методом дослідження за допомогою FISH (заключний діагноз). Останній виконується в спеціалізованому медико-генетичному центрі на кафедрі медичної генетики КМАПО ім. П.Л. Шупика.
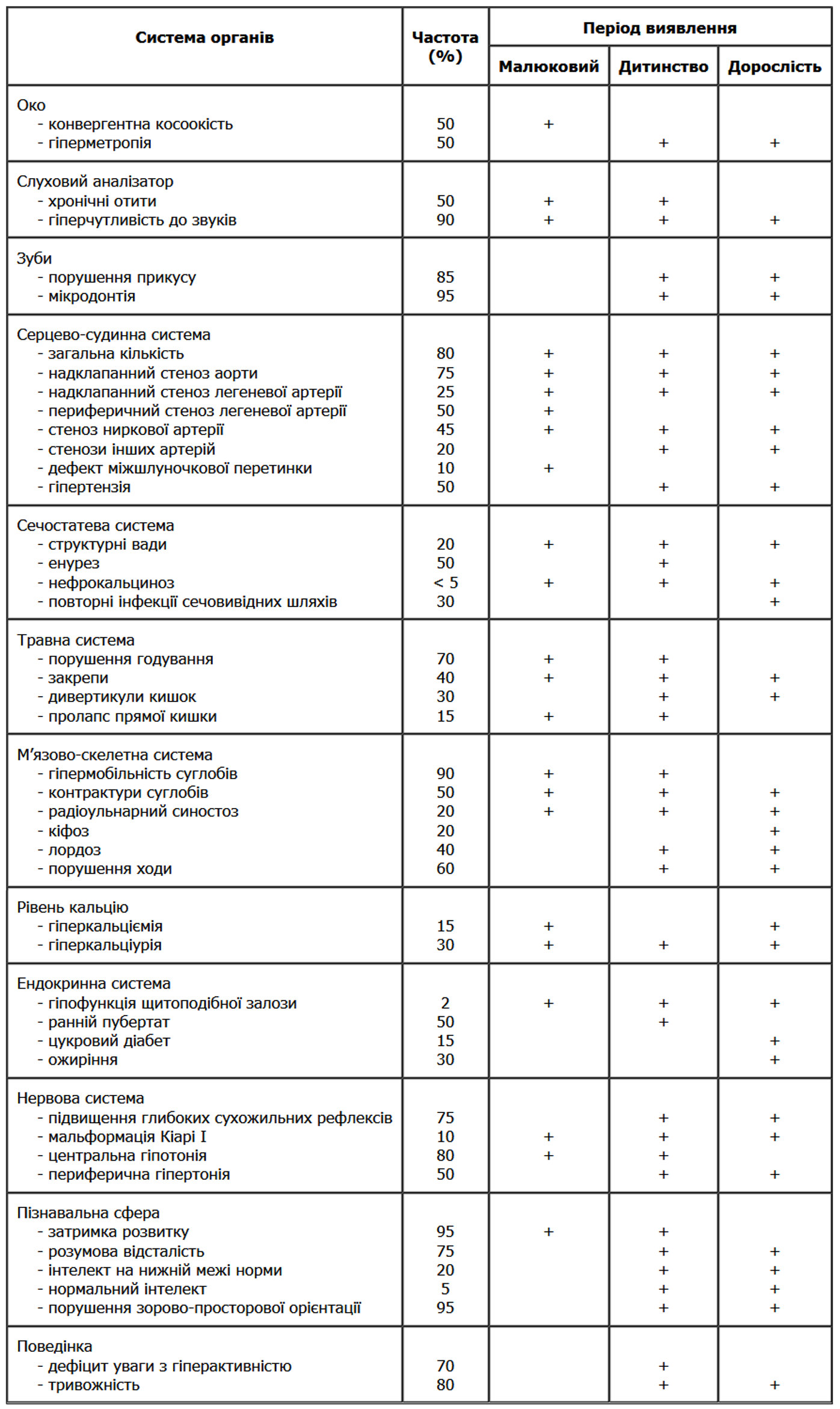
1 етап. Запідозрити наявність синдрому Вільямса повинен лікар першого контакту (педіатр, лікар загальної практики, кардіолог, кардіохірург). Перелік клінічних симптомів, які можуть виявлятися у хворих із синдромом Вільямса, представлений у таблиці 2.
Таблиця 2. Структурні та функціональні порушення при синдромі Вільямса
(з рекомендацій Американської педіатричної асоціації)
При підозрі на наявність у пацієнта синдрому Вільямса необхідно проводити наступний перелік обстежень:
- повне фізичне та неврологічне обстеження;
- кардіологічне обстеження з вимірюванням артеріального тиску на всіх кінцівках окремо, та Ехо-КГ;
- УЗД нирок та органів малого тазу;
- дослідження функції нирок (рівень азоту та креатиніну крові);
- уріналізис;
- дослідження рівню кальцію крові та сечі;
- дослідження функції щитоподібної залози;
- офтальмологічне дослідження;
- оцінку психомоторного розвитку.
2 етап. Встановлення клінічного діагнозу лікарем генетиком в обласному медико-генетичному центрі, за направленням лікаря першого контакту. Хоча деякі симптоми синдрому Вільямса мають місце вже при народженні, повний спектр малих аномалій обличчя, що складає враження “обличчя ельфа” та дозволяє поставити клінічний діагноз, як було вже зазначено вище, формується на 3-4-му році життя. Для ранньої діагностики основне значення має оцінка клініко-лабораторних змін. Бальна шкала полегшує встановлення клінічного діагнозу (таблиця 3). При огляді пацієнта в таблиці відмічають наявність симптомів. По кожному розділу підраховують кількість пунктів, що відмічено, та виставляють відповідну кількість балів з визначенням загальної суми.
Таблиця 3. Бальна шкала для клінічної діагностики синдрому Вільямса

В ранньому віці FISH дослідження слід проводити всім дітям з надклапанним стенозом аорти або периферійним стенозом легеневої артерії, незважаючи на відсутність характерного обличчя та психо-когнітивного профілю.
3 етап. Підтвердження клінічного діагнозу синдрому Вільямса за допомогою FISH в спеціалізованому медико-генетичному центрі. Попередньо рекомендується проведення прометафазного цитогенетичного дослідження для виключення складних цитогенетичних перебудов з участю хромосоми 7.
FISH-метод – це вид молекулярної гібридизації нуклеїнових кислот, при якій одним компонентом гібридизації є ДНК фіксованого матеріалу (ДНК про- та метафазних хромосом, а також ДНК інтерфазних ядер), а іншим компонентом – комплементарна їй послідовність ДНК в розчині, помічена флуорохромами (ДНК-зонди). Для діагностики синдрому Вільямса застосовують локус–специфічний ДНК зонд Williams Syndrome Region Probe. Мікроделеція 7q11.2 виявляється у 99% пацієнтів, які мають специфічні клінічні ознаки. Матеріалом для дослідження є периферійна кров пацієнта, взята в стерильний моновет з гепарином.
Лікування:
Як було зазначено вище, причиною розвитку синдрому є мікроделеція, тобто втрата ділянки хромосоми 7. Ця подія відбувається під час кросинговеру в клітинах батьків, і фактори, що її викликають, рівно як і фактори, що дозволять цьому запобігти, на сучасному етапі ще не з’ясовані. Тому, як і для інших хромосомних синдромів етіологічного та патогенетичного лікування синдрому Вільямса на сьогоднішній день не існує.
В той же час адекватне симптоматичне лікування, яке поєднує медикаментозну терапію, хірургічну корекцію вроджених вад розвитку та заходи психо–педагогічної корекції, дозволяє суттєво покращити вітальний та медичний прогноз та досягти значної соціальної адаптації пацієнта в суспільстві.
Діти з синдромом Вільямса потребують проведення ранніх загальнооздоровчих та лікувально–корекційних заходів, які в ранньому віці доповнюються обмеженням вживання вітаміну Д. Рекомендується виключити прийом мультивітамінних комплексів, у зв’язку з тим, що кожен з них включає вітамін Д. Крім того, в деяких випадках, слід обмежити перебування дитини на сонці, для зменшення вироблення вітаміну Д в шкірі під впливом УФ випромінювання. Гіперкальціємія може потребувати в деяких пацієнтів зменшення вживання кальцію з їжею, при цьому важливо проводити моніторинг рівню кальцію, щоб уникнути розвитку ятрогенного рахіту.
Лікувальні заходи в перші роки життя скеровані насамперед на корекцію серцево-судинних порушень та стабілізацію обмінних процесів. Хірургічної корекції надклапанного стенозу аорти потребують приблизно 30% пацієнтів. Летальність при проведенні кардіохірургічного втручання складає 3-7%. Всім хворим з синдромом Вільямса необхідно проводити регулярне вимірювання артеріального тиску окремо на верхніх та нижніх кінцівках (щорічно) та Ехо-КГ (кожні 3-4 роки). Регулярні обстеження необхідні навіть при нормальних базових показниках.
Як було зазначено вище, більшість пацієнтів з синдромом Вільямса мають затримку росту, а кінцеві ростові показники зазвичай нижче 3%. Важливим є контроль росту дітей за таблицями згідно спеціально розробленим стандартам. У кожному випадку затримки росту проводиться визначення рівню гормонів щитоподібної залози та гормону росту з подальшою їх корекцією при необхідності.
Підвищеної уваги та інтенсивного лікування може потребувати хронічне запалення середнього вуха. Порушення прикусу обумовлюють необхідність проведення ортодонтичної корекції.
В зв’язку з підвищеною збудливістю пацієнта необхідність застосовування наркозу виникає не лише при проведенні будь-яких хірургічних маніпуляцій, а навіть, в деяких випадках, при ультразвуковому дослідженні. Необхідно пам’ятати про підвищений ризик раптової смерті при проведенні наркозу у дітей з синдромом Вільямса. Застосування наркозу у таких дітей можливо тільки за участю досвідченого анестезіолога та з забезпеченням можливості проведення термінових реанімаційних заходів.
Особливо важливе значення має рання стимуляція психомоторного розвитку дитини та корекція порушених рухових функцій. У зв’язку з цим показані загальнозміцнюючий масаж та лікувальна фізкультура, що направлені на стимуляцію розвитку статичних та локомоторних функцій. Необхідні також спеціальні вправи, які розвивають тонку моторику пальців рук та зорово-моторну координацію. Необхідно приділяти увагу корекції косоокості – використовуються лінзи, іноді потрібне хірургічне втручання.
Протягом раннього, дошкільного та шкільного віку необхідно стимулювати у дітей пізнавальну активність, розвивати вміння чітко діяти за інструкцією дорослого. Велику увагу під час логопедичних занять слід приділяти розвитку змістової складової мови, при цьому необхідно навчати дитину уникати механічного повторювання та мовної активності, яка позбавлена конкретного змісту. Розробляючи програму занять слід враховувати гарні здібності дітей з синдромом Вільямса до засвоювання навиків читання.
Диспансерний нагляд:
Медичне обслуговування пацієнтів з синдромом Вільямса потребує розуміння динаміки розвитку цього спадкового захворювання, знання потенційно можливих ускладнень та проведення періодичних медичних оглядів. Внаслідок варіабельності клінічних проявів діагноз синдрому може бути не встановлено в ранньому дитячому віці. Слід ще раз наголосити, що клінічний діагноз, встановлений в будь-якому віці потребує підтвердження проведенням FISH, з виявленням мікроделеції 7q11.23.
План диспансеризації хворого з синдромом Вільямса:
- Регулярне медичне обстеження з контролем росто-вагових показників, гостроти зору та слуху.
- Моніторинг рівню артеріального тиску, пульсу на стегновій артерії та щорічне кардіологічне обстеження з проведенням Ехо-КГ (до п’яти років, при нормальних показниках наступне Ехо-КГ на початку пубертату).
- Щорічне проведення сечового скринінгу (уріналізис).
- Контроль рівню кальцію крові – щорічно, якщо в анамнезі були епізоди гіперкальціємії. Якщо первинні показники були в межах норми проведення дослідження кожні 3-4 роки.
- Рівень екскреції кальцію з сечею – дослідження кожні 2 роки.
- Функція щитоподібної залози – обстеження з визначенням рівню гормонів кожні 4 роки.
- Корекція порушень годування в ранньому віці (рефлюксна хвороба, закреп).
- Обстеження іншими фахівцями при наявності показань (ортопед, дитячий невролог, гастроентеролог, нефролог та ін.).
- Ранній початок психо–педагогічних заходів, занять з логопедом, розробка індивідуальної освітньої програми.
Важливим моментом в роботі з родиною, що має дитину з синдромом Вільямса, є забезпечення доступу до друкованої інформації, стосовно синдрому, та інформації в мережі Internet, скерування до відповідних батьківських асоціацій.
Завдання медико-генетичного консультування: 1) індивідуальна оцінка клінічних симптомів та встановлення клінічного діагнозу; 2) визначення ризику повторного народження хворої дитини в родині та ризику для інших родичів; 3) обговорення можливостей реалізації репродуктивної функції у дитини з синдромом Вільямса, 4) обговорення можливих ускладнень з батьками.
Більшість випадків синдрому Вільямса є спорадичними, хоча описано декілька родинних випадків синдрому з успадкуванням хвороби як від матері, так і від батька, причому у всіх цих випадках батьки мали фенотипові ознаки синдрому Вільямса у вигляді особливостей обличчя та інтелектуальної недостатності. Ризик для сибсів при мікроделеції de novo не перевищує 0,5%, ризик для нащадків складає 50%. Репродуктивна функція у пацієнтів з синдромом Вільямса зазвичай не порушена, обмеженням для її реалізації може бути важка ступінь розумової відсталості, або соціальна неадаптованість хворого. В будь-якому випадку батьки або опікуни пацієнта повинні бути попереджені про високий генетичний ризик для нащадків при синдромі Вільямса та необхідність проведення пренатальної діагностики.
Перелік рекомендованої літератури:
- Мостюкова Е.М., Мостовкина А.Г. Основы генетики: Клинико–генетические основы коррекционной педагогики и специальной психологии. Учеб. пособие для студ. пед. высш. учебн. заведений – М.: Гуманит.изд.центр ВЛАДОС, 2001.- 368 с.
- Наследственные синдромы и медико-генетическое консультирование. Атлас-справочник /С.И. Козлова, Н.С. Демикова, Е.Семанова, О.Е.Блинникова.- М.: Практика, 1998.- 416 стр., 392 ил.
- Ворсанова С.Г., Юров Ю.Б., Чернышов В.Н. Хромосомные синдромы и аномалии. Классификация и номенклатура.- Ростов-на-Дону, 1999.- 191 с.
- Бочков Н.П. Клиническая генетика. Учебник для вузов.- М.: ГЭОТАР-МЕД, 2001.- 448 с.
- American Academy of Pediatrics: Health care supervision for children with Williams syndrome.Pediatrics 2001 May;107(5):1192-204.
- Morris CA. Williams syndrome. Gene Clinics: Medical Genetics Knowledge Base (http://www.geneclinics.org).
Переглянуто редакційною колегією I.B.I.S.: 28/10/2005
Дивіться також:
- Синдром Вільямса (інформація для спеціалістів)
- Синдром Вільямса (інформація для батьків)
- Синдром Вільямса (діаграми розвитку дітей)
|
|
|
|
|
|




